近日,中国科学院微生物研究所研究员王军团队在Nature Communications(IF=14.919)上发表了题为"Short- and long-read metagenomics expand individualized structural variations in gut microbiomes"的论文。

中国科学院微生物研究所王军研究员和动物所宋默识研究员为共同通讯作者;中国科学院微生物研究所助理研究员陈亮和赵娜,博士研究生曹家宝,硕士研究生刘小林等为第一作者;上海百趣代谢组学技术研究中心创始团队刘志鹏研究员和研究人员范艳群为论文共同作者。
该研究建立了ONT三代测序和Illumina二代测序数据混合组装的新方法(图1a),检测出了更多包括插入突变、缺失突变和基因倒位在内的结构变异(structural variations, SVs)。同时,通过对100个人组成的健康人群横断面队列和由10个人组成的纵向跟踪队列的宏基因组学和代谢组学的联合分析,发现了SVs在不同个体间存在明显不同,但在同一个体内有相对稳定,同时也发现SVs不仅影响菌群与代谢物的功能,对人体表型也有一定影响。
研究团队首先用已知数据集(ZymoBIOMICS™ Microbial Community)对ONT和Illumina的混合组装方法与其他几种组装方式进行比较,发现混合组装方式从完整度、污染率、细菌基因组平均遗传相似度(average nucleotide identity, ANI)和编码密度方面都有更好的效果。同时通过对两个人群的肠道数据分析发现,混合组装方式能提高数据质量。与二代宏基因组组装结果比较发现,混合组装方式虽然少了17.3%的contigs,但组装序列数量多了5.1%,N50值提高了3倍多。对contigs进行分箱后得到能代表单个菌种的重建宏基因组装基因组
(metagenome-assembled genomes,MAGs),通过混合组装方式得到平均N50为117kb的9,612个MAGs(每个样本20~83个),去重后得到692个MAGs(图1b,1c),其中有623在UHGG数据库中,且有208个MAGs的质量更高,同时也发现了67个新的genomic bins,用新版本dRep去重后减少了2个MAGs。从全面性考虑,159个非冗余的MAGs均包含了23S、16S和5S rRNA序列,448个MAGs至少含有其中一种类型的rRNA。基于Illumina的组装方式得到616个MAGs,N50约为混合组装的一半,且只有9个MAGs含有三种类型的rRNA序列,258个MAGs含有至少一种rRNA序列。所有样本中,Fusicatenibacter saccharivorans出现的频率最高,其次是Anaerostipes hadrus和gathobacter
rectalis,有189个菌以MAGs的形式出现在至少10个样本中。
鉴于ONT测序能发现更多SVs的特点,通过MAGs的比对,发现多种类型的SVs。189个菌通过dRep比对,鉴定出了317558个插入突变,34129个缺失突变和1373个基因倒位(图1d);接下来又随机选取插入突变和缺失突变两个峰中(140~160bp和1050~1150bp,图1e)SVs片段进行分析,发现移动元件和染色体外移动基因元件(extrachromosomal mobile genetic elements,eMGEs)在两种突变的短SVs片段中更多,从而推断短序列的SVs可能与噬菌体整合和其他移动元件相关;但并不是所有SVs都有可检测的移动元件,其他的SVs可能是复制和重组引起,具体机制有待进一步验证。
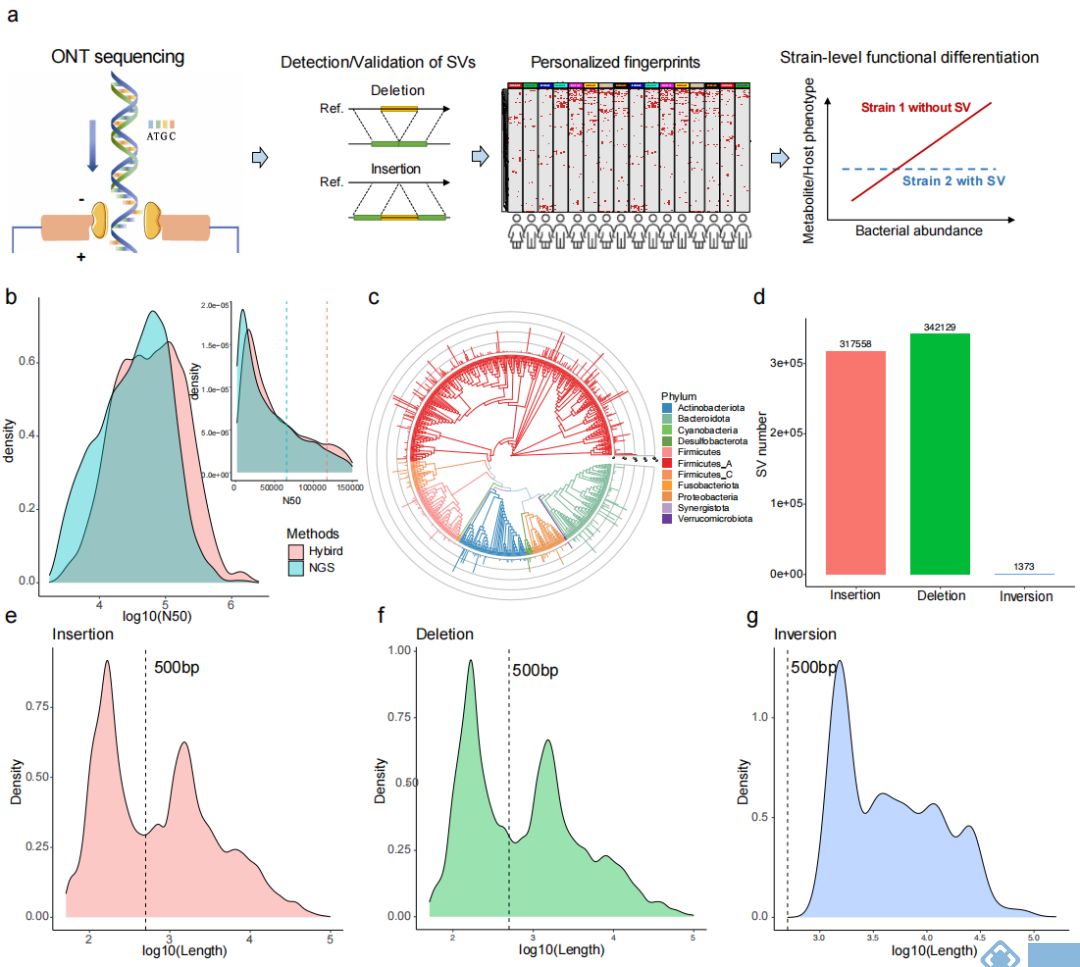
图1. 三代和二代混合组装方式验证结果
接下来,通过重新匹配参考MAG或者MAG中含有SV的序列,以进一步验证检测出的SVs。人工匹配后发现97%以上随机挑选SVs集与ONT多处位置的Reads数目一致,从而验证了单分子测序得到特异SVs的可靠性(图2a),同时也发现同一个体相同细菌基因SVs的低异质性。
对种水平(MAGs)的SVs分析发现,SVs总数与所有样本中的MAGs数和基因大小正相关。但由于细菌基因组中SVs在人群中分布的不均匀性,所以进一步校正平均SV数和基因组大小,发现1M基因组中门水平多样性最高的Firmicutes有20.4的SVs,Akkermensia所属的Verrucomicrobia有19.5的SVs,而Desulfobacteroita和Proteobacteria的SVs最少(图2b,2c)。
对两个人群的189个MAGs分析发现,不同个体间每Mb基因组中有16.7的SVs,而同一个体不同时间点每Mb基因组种SVs的中位值为0。因此,SVs可用于区分个体间的细菌种类和肠道菌群,同时对于特定菌10天内在个体内的稳定性(图2d)结果间接表明LifeLines cohort队列发现的3年内菌株分化或置换可能是由于SV的逐步累积引起。
 、
、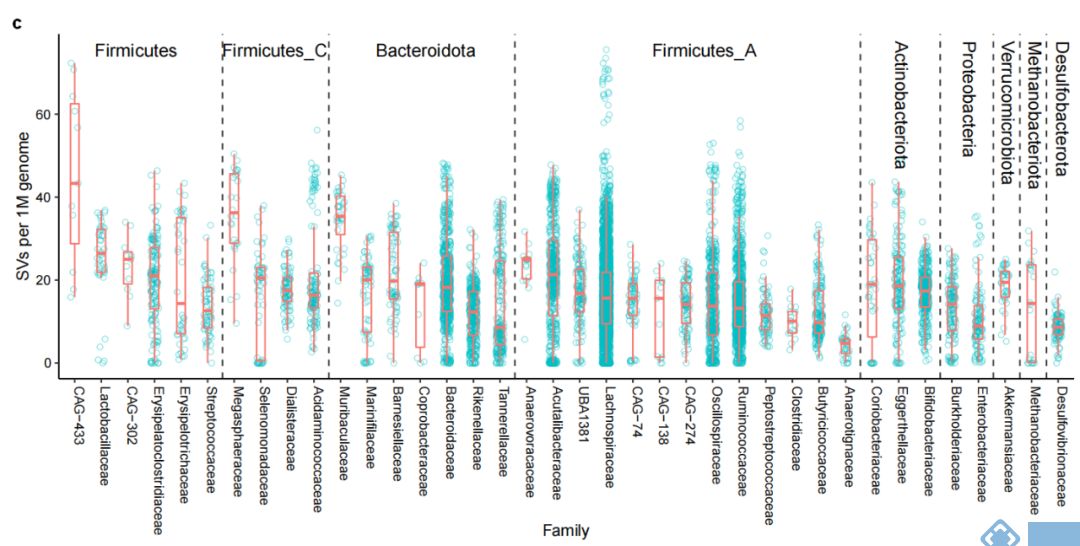
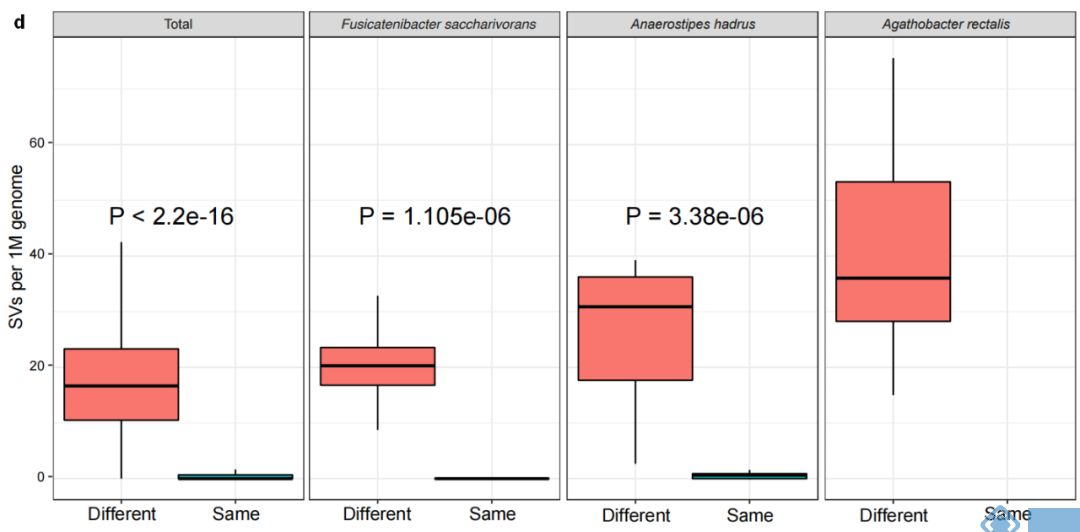
图2. 人体肠道微生物结构变异结果
接下来,对人群中检测出来的SVs相关的基因进行功能富集分析,发现267个通路与插入突变和缺失突变(图3a)相关,但未发现与基因倒位相关的通路,前30个通路中有19个通路是与多糖降解、鞘脂代谢组学等与代谢组学相关的通路;同时也发现一些与环境信息处理相关的通路(如磷酸转移酶系统(phosphotransferase system,PTS)和ABC转运蛋白等)。
为进一步研究SVs对机体功能(尤其是微生物代谢组学)的影响,对横断面队列的粪便、血清和尿液样本进行代谢组学分析,结果表明,SVs导致基因功能发生改变,从而使得SVs突变组中的菌与代谢物不相关,而不含SVs突变组中菌与代谢物显著相关。相关性分析表明,11个菌与粪便、血清和尿液中的代谢物显著相关,其中涉及到889个受SV影响的细菌-代谢物关联对(图3b,3c)。
SVs与代谢组学的关联分析发现,70个SVs影响了细菌与74个粪便代谢物显著性关联,31个SVs影响了细菌与66个尿液代谢物的关联,2个SVs影响了细菌与 2个血清代谢物显著关联。之前的研究中,inositol已被发现与Anaerostipes hadrus的缺失突变有关,而本文研究中发现Bacteroides uniformis基因组的基因座上插入突变和缺失突变均使得该菌与尿液样本中inositol的关联消失。12个SV-affected基因的存在,使得Fusicatenibacter saccharivorans与粪便样本中Neotrehalose的关联不显著(图3d);同样,33个SV-affected基因的存在使得Agathobacter rectalis与F1P间的关联不显著(图3e)。功能分析的结果也表明SVs通过影响相关基因的功能对菌和代谢物关联产生影响。
为进一步研究SVs突变对表型的影响,选取横截面队列样本中受SVs影响的两个代谢物F1P和neotrehalose与空腹血糖做关联分析,发现F1P和neotrehalose均与空腹血糖显著负相关,且F.saccharivorans与空腹血糖也显著负相关,但在SVs亚组中,关联变得不显著(图3h);SVs的存在也使得A.rectalis与glucose的关联减弱(图3i)。
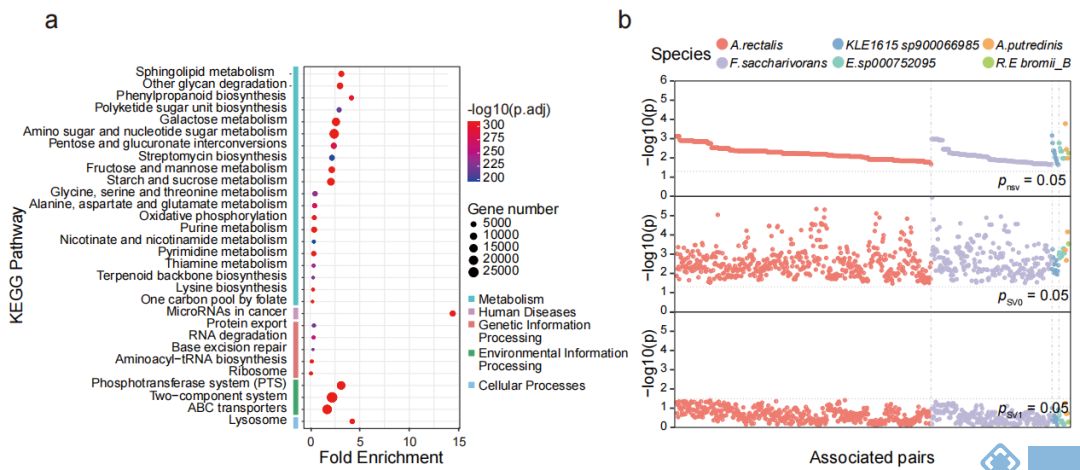
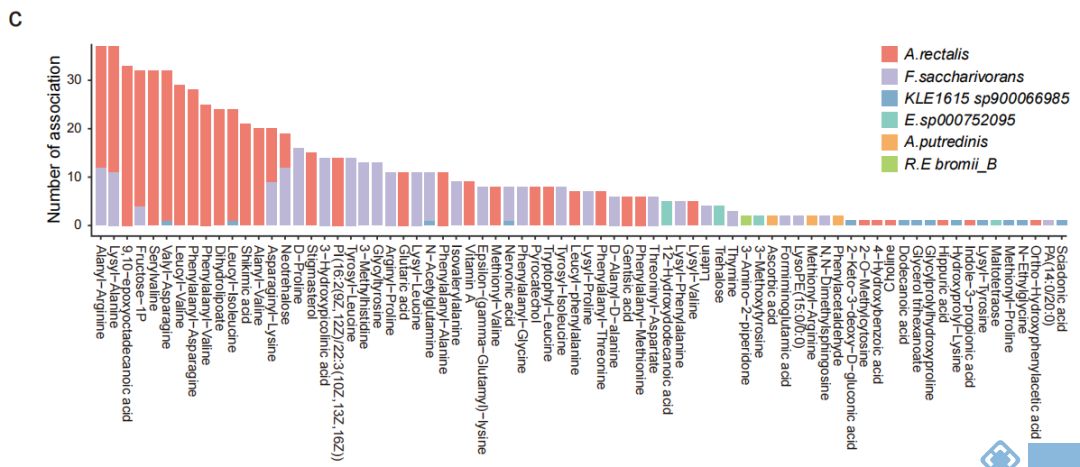
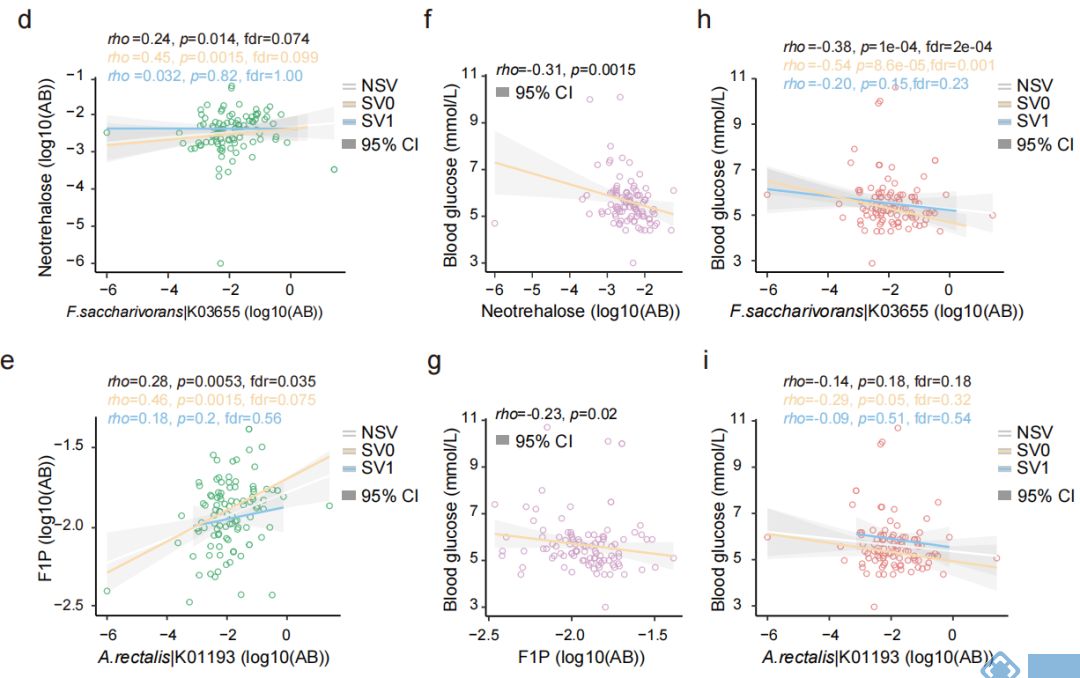
图3. 肠道微生物中与SVs相关的功能研究结果
由于噬菌体侵染细菌基因组和病毒的逃离均会导致SVs的产生,因此用ProphageHunter对所有MAGs进行分析,得到基因组大小在1236bp和91792bp间以长尾噬菌体Siphoviridae和肌尾噬菌体Myoviridae为主的2247个原噬菌体(图4a)。对原噬菌体元件和细菌基因组进行关联分析,得到1,077个原噬菌体-宿主对(图4b);其中,只有72个在MVP数据库中;而二代测序数据只检测到1815个原噬菌体,其中80.77%在混合组装中检测到;从结果我们可以看出,ONT-二代混合组装数据更有利于原噬菌体的发现。
除原噬菌体外,菌群基因中还有用于抵抗病毒重复感染的CRISPR-Cas系统,该系统中loci的spacers包含有特定病毒的特征序列,可能与菌种的插入突变或者缺失突变有关。同样,对所有MAGs的分析发现了150058个CRISPR
spacers,平均每个样本中1665±560个spacers,大部分的spacers是新发现的,只有17,600个(11.73%)在CRISPROpenDB数据库汇总,22962(15.30%)在西方人群的肠道菌群中出现;基于二代测序的组装方式,只发现了9542个spacers。由此我们也能看出,新的宏基因组组装方式具有更强的发现基因元件(如CRISPR spacers)的能力。
对原噬菌体/CRISPR spacers的β多样性分析发现(Jaccard distance距离),横截面队列中个体的差异性显著大于跟踪队列个体内的差异性。群体水平对原噬菌体和CRISPR spacers的组成分析表明两者间有较强的共变;能揭示原噬菌体和病毒群落组成间相关性的Procrustes分析结果表明,横截面队列中不同个体间原噬菌体和病毒组成显著相关(图4c);对宏基因组数据中活性病毒序列的分析发现,2247个鉴定出的原噬菌体汇中有47个有潜在活性的,从而表明细菌基因中存在大量无活性的原噬菌体,从而保持SVs的稳定性。
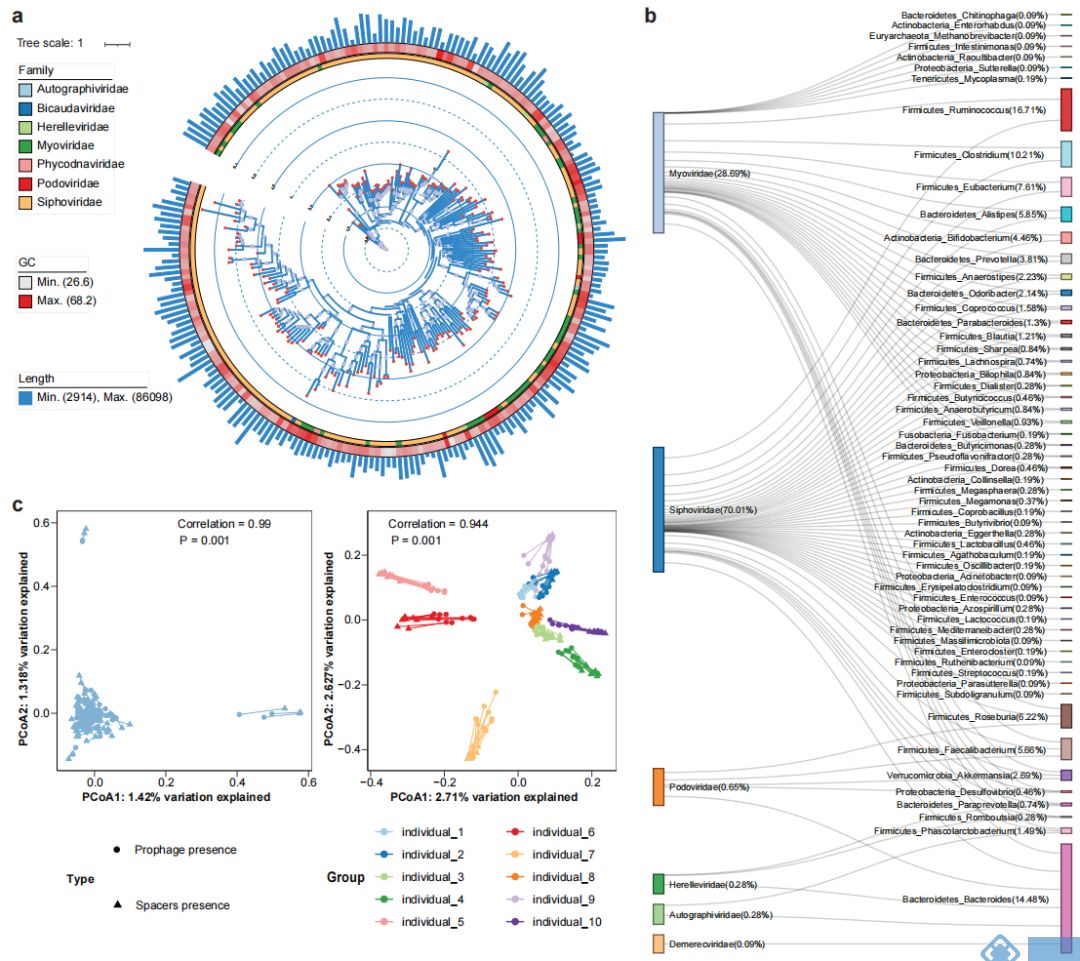
图4. 肠道菌群汇中与病毒和CRISPR相关的研究结果
本研究建立了基于三代测序和二代测序的混合组装方式,不仅提高了数据质量,也能检测出大量包括插入突变和基因倒位在内的结构变异,也有利于原噬菌体以及CRISPR spacers等基因元件的发现。同时通过横截面队列和纵向跟踪队列数据的分析,发现SVs在不同个体间存在较强的异质性以及个体内的稳定性;通过功能分析和代谢组学分析,发现SVs能影响菌群与代谢物和表型间的关联。
文/阿趣代谢组学